OAS1 splicing found to be associated with risk of severe COVID-19
Published: 2022-02-09

Several large-scale international efforts, for example the COVID-19 Host Genetics Initiative, are today working to understand the importance of genetic factors in COVID-19 susceptibility and severity. A number of genomic regions have been associated with severe COVID-19, but more research is needed to identify the causal agents within those regions, and further our knowledge of COVID-19 pathophysiology.
Previous research (Pääbo & Zeberg 2020; Pääbo & Zeberg 2021) has identified two risk locus associated with the relative risk of developing severe COVID-19; one on chromosome 3 (3p21.31), and the other at the OAS1/2/3 locus on chromosome 12 (12q24.13). The haplotypes in question are both of Neanderthal origin (more information about decoding the Neanderthal genome, can be found here. The connection to Neanderthals adds to the complexity of finding the casual variants at the locus. Both Neanderthal and Denisovan haplotypes are typically very long, and linkage disequilibrium (LD) further increases the difficulty of identifying such variants. Recent research identified a protective haplotype at the OAS1/2/3 locus (about 75 kb in length, spanning OAS1, OAS2, and OAS3 genes) that is common in individuals with European ancestry. In addition, rs10774671 (OAS1) has been proposed as a candidate causal gene-variant, but further research into disentangling the association to severe COVID-19 is warranted.
A recent article (Jan 14th 2022) used multi-ancestry fine mapping to study the OAS1 splicing and elucidate how it is involved in the risk of severe COVID-19. The study involved an international collaboration of researchers from the US, Canada, UK, Japan, Finland, Germany, and Sweden (first author Jennifer E. Huffman (Massachusetts Veterans Epidemiology Research and Information Center, Boston), and corresponding author Hugo Zeberg (Department of Neuroscience, Karolinska Institutet, and Max Planck Institute for Evolutionary Anthropology, Leipzig).
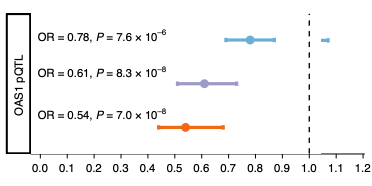
Huffman and colleagues tested associations in different ancestry groups known to have different LD structures and shorter haplotypes to elucidate which SNPs at the rs10774671 affect the severity of COVID-19. Using 1000 Genomes Project, the researchers showed that several variants co-segregate with rs10774671 in individuals with South Asian or East Asian ancestry (129 and 128 variants, respectively), but not in individuals with African ancestry (LD r2 > 0.6). In addition, Neanderthal haplotypes are generally not detectable among individuals of primarily African ancestry. The researchers therefore selected individuals of primarily African ancestry to determine whether rs10774671 is associated with COVID-19 severity. The researchers used multi-ancestry fine mapping to examine the association between the splice acceptor variant rs10774671 and the outcomes in patients of primarily African ancestry in six studies (total N=2,787 cases, and N=130,997 controls). Notably, the results showed a splice variant of OAS1 in people of African ancestry that was independent of gene flow from Neanderthals. The rs10774671 G allele was found to protect against COVID-19 hospitalization in individuals of either African (OR = 0.94; 95% CI = 0.88–0.99) or European ancestry (OR = 0.92, 95% CI = 0.90–0.95) (P = 0.03). The included studies were not found to exhibit statistical differences in the cohorts, nor exhibiting other methodological large differences, that could explain this. The results were confirmed by the COVID-19 Host Initiative using a meta-analysis of individuals of African ancestry. These results showed that the rs10774671 G allele confers protection against severe COVID-19, which is not dependent on the variants with which it is associated in non-African populations.
In summary, using trans-ancestry fine-mapping, the researchers studied 20,779 hospitalized cases, and demonstrated that the rs10774671 G allele is likely protective against severe COVID-19. This strongly indicates that OAS1 is an effector gene, responsible for influencing COVID-19 severity. The researchers suggest that future genetic association studies include populations with different ancestries, and highlight the importance of rapidly sharing data through large, international consortia.
‘This study shows how important it is to include individuals of different ancestries. If we had only studied one group, we would not have been successful in this case’ says Hugo Zeberg.
Funding
A number of international funders supported this study, as well as the Jeanssons and Magnus Bergvalls Foundations and the Swedish Research Council
Data
- COVID-19 summary statistics for individuals of African ancestry
- CADD-scores v.1.6
- Genomes from the 1000 Genomes Project
- The fine-mapping association summary statistics
- The code used for the fine-mapping analysis
- Source data are provided with this paper
Article
DOI: 10.1038/s41588-021-00996-8
Huffman, J.E., Butler-Laporte, G., Khan, A. et al. Multi-ancestry fine mapping implicates OAS1 splicing in risk of severe COVID-19. Nat Genet (2022).